FRAP experiments
Objectives
Here, we want to characterize the dynamic of the endoplasmic reticulum in living cells using a FRAP approach.
Material
A Nikon Ti2 Eclipse microscope (Nikon, Tokyo, Japan) with a Plan Apo λ 60x 1.4 NA oil immersion objective (MRD01605; Nikon) was used. For this experiment, the camera was a digital CMOS ORCA-Fusion BT (C15440-20UP; Hamamatsu Photonics, Hamamatsu, Japan) and the light engine was from CoolLED (pE-800fura; CoolLED, Andover, United Kingdom). Videomicroscopy and FRAP were performed using Inscoper scanFRAP (INSCOPER, Cesson-Sévigné, France) with a 405nm laser source (L6Cc; Oxxius, Lannion, France). All images were deconvolved using Microvolution software (Microvolution, Cupertino, CA, USA).
Method
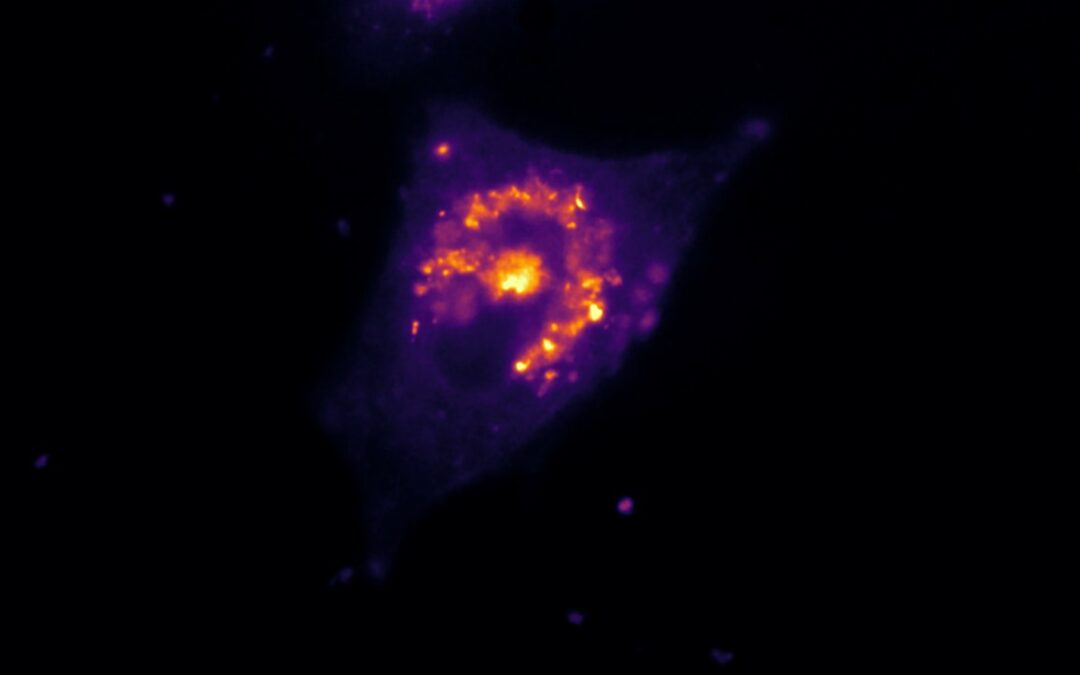
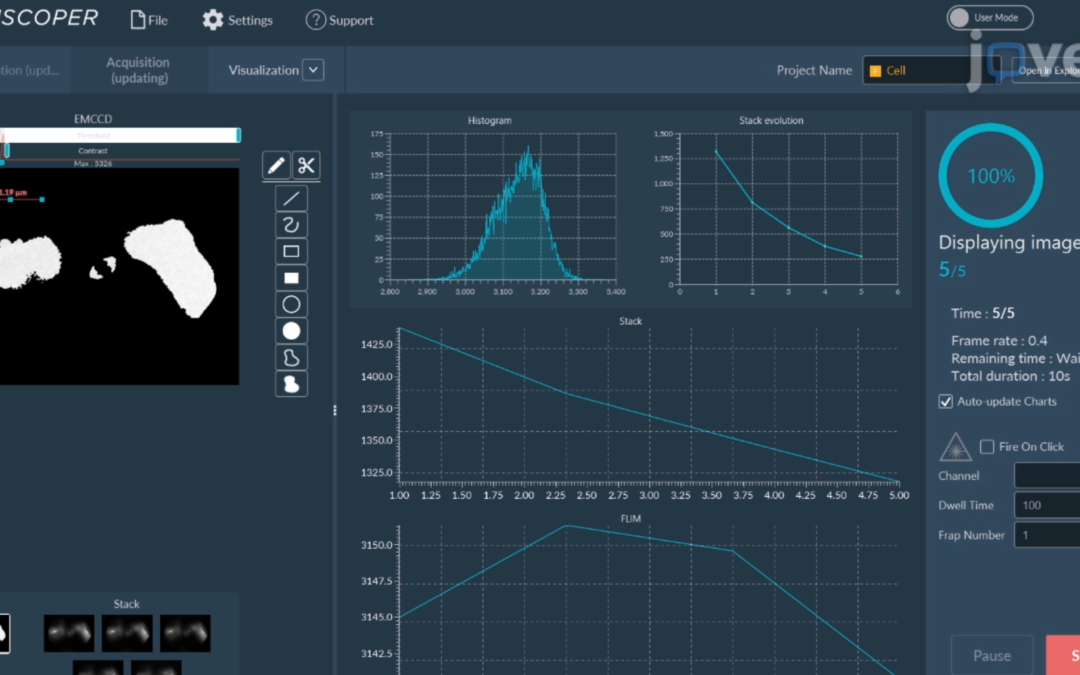
HeLa cells were transfected to overexpress YFP (Yellow Fluorescent Protein) in their endoplasmic reticulum. For the FRAP experiment, ROI was defined and bleached using the 405nm laser on living cells and fixed cells as negative control. The fluorescence recovery was then measured. FRAP experiments are basically composed of three steps. First, a short pre-bleached timelapse is captured to get basal levels of fluorescence. Here, a stable and strong YFP signal was observed in transfected cells before photomanipulation. Then, the region of interest (ROI) is selected and photobleached with the appropriate laser settings (wavelength, intensity, point density, …). Finally, another timelapse is performed to monitor in real-time the fluorescence recovery in the bleached ROI.
Results
The laser microirradiation represents a powerful tool to monitor DNA repair with high temporal and spatial resolution. DNA lesions were induced following ROI (region of interest) previously defined using the Inscoper software. During the acquisitions, a progressive recruitment of ALC1-GFP protein could be observed over the entire length of the ROI (Figure 3A). Mean intensity of the fluorescent signal has been then measured on the damaged area. The signal from an unaltered ROI was also measured as control. A rapid increase of the signal was observed in the irradiated area, to remain stable in less than 10 seconds (Figure 3B). On the contrary, intensity from the control ROI appeared slightly decreased following the photomanipulation. ALC1-GFP accumulation could also be characterized by the width of the band (Figure 3C). In this experiment, the phenomenon was rapid with a half time of 47.7 ± 7.1 seconds and total recovery in just over 300 seconds (Figure 3D). YFP-labeled proteins from the ER in living cells appeared to be mobile and able to rapidly diffuse inside the cell cytoplasm.